製薬会社の拡大を計画していますか? インドへの事業設立 しかし、どこから始めればよいかわからない?インドは最も急成長している医薬品市場の1つであり、コスト面で有利であり、熟練した専門家が多数存在しています。2023~24年度にかけて、インドの医薬品輸出はほぼ達成しました。 280億米ドル、米国だけで約 31%。製薬事業をインドにアウトソーシングすることで、コストを削減することもできます。 60— 75%、米国企業の製造、研究開発、およびサポートサービスの経費削減を支援します。
このブログでは、コンプライアンス要件、登録、ライセンス、アウトソーシングの機会、およびVJM Globalのサポート方法など、インドで製薬会社を設立する方法をご案内します。 米国企業 プロセス全体を通して。
始める前に
- 正しい事業構造を選択し、FDIルートを確認することが、長期的な事業運営を形作る第一歩です。
- インドのコンプライアンスには、スケジュールMの製造基準から税務申告やデータ保護義務に至るまで、継続的な注意が必要です。
- 簿記、給与計算、監査サポートなどのアウトソーシング機能により、製品開発とライセンス管理にリソースを集中させることができます。
- ほとんどの医薬品免許では名前を記録する必要があるため、最初に資格のある薬剤師と技術スタッフを任命することが必須です。
- とのパートナーシップ VMグローバル 米国とインドの規制に関する二重の専門知識を提供し、コスト管理をサポートしながらリスクを軽減します。
米国企業が製薬事業拡大のためにインドを検討すべき理由
インドは、特にジェネリック医薬品を中心に、世界の医薬品サプライチェーンにおける重要な拠点としての地位を確立しています。以下について供給しています。 20% 世界のジェネリック医薬品の量別および総数 40% 米国のジェネリック医薬品需要の
米国の製薬企業家にとって、インドには3つの大きな利点があります。
- 専門性の高い大規模な人材プールへのアクセス
薬事務、製剤開発、品質保証の分野で熟練した専門家が幅広く在籍しています。たとえば、米国食品医薬品局(FDA)への提出書類の準備をしている場合、インドには米国のコンプライアンス要件を満たす書類作成の経験があるチーム(ANDA、DMF)があります。
- 品質を犠牲にすることなくコストを削減
会計、コンプライアンス監視、または二次製造活動をインドにアウトソーシングすることで、経費を大幅に削減できます。多くの米国企業はすでにインドの委託研究機関(CRO)に臨床試験管理を委託しています。これは、同じ試験を国内で実施する場合に比べてコストはわずかでありながら、国際的なGCP基準を満たしているためです。
- コンプライアンスに裏打ちされた信頼性の高いインフラストラクチャ
約500のFDA承認施設があるインドでは、生産環境と試験環境が米国の期待に合っていることを安心させてくれます。これが、米国で調剤される処方箋の10分の4がインドの製造業者によって供給されている理由です。これは、アメリカの医療がすでにインドの医療能力に依存していることを示す一例です。
インドの利点を理解したら、次のステップは製薬会社を設立するための正確なプロセスを知ることです。
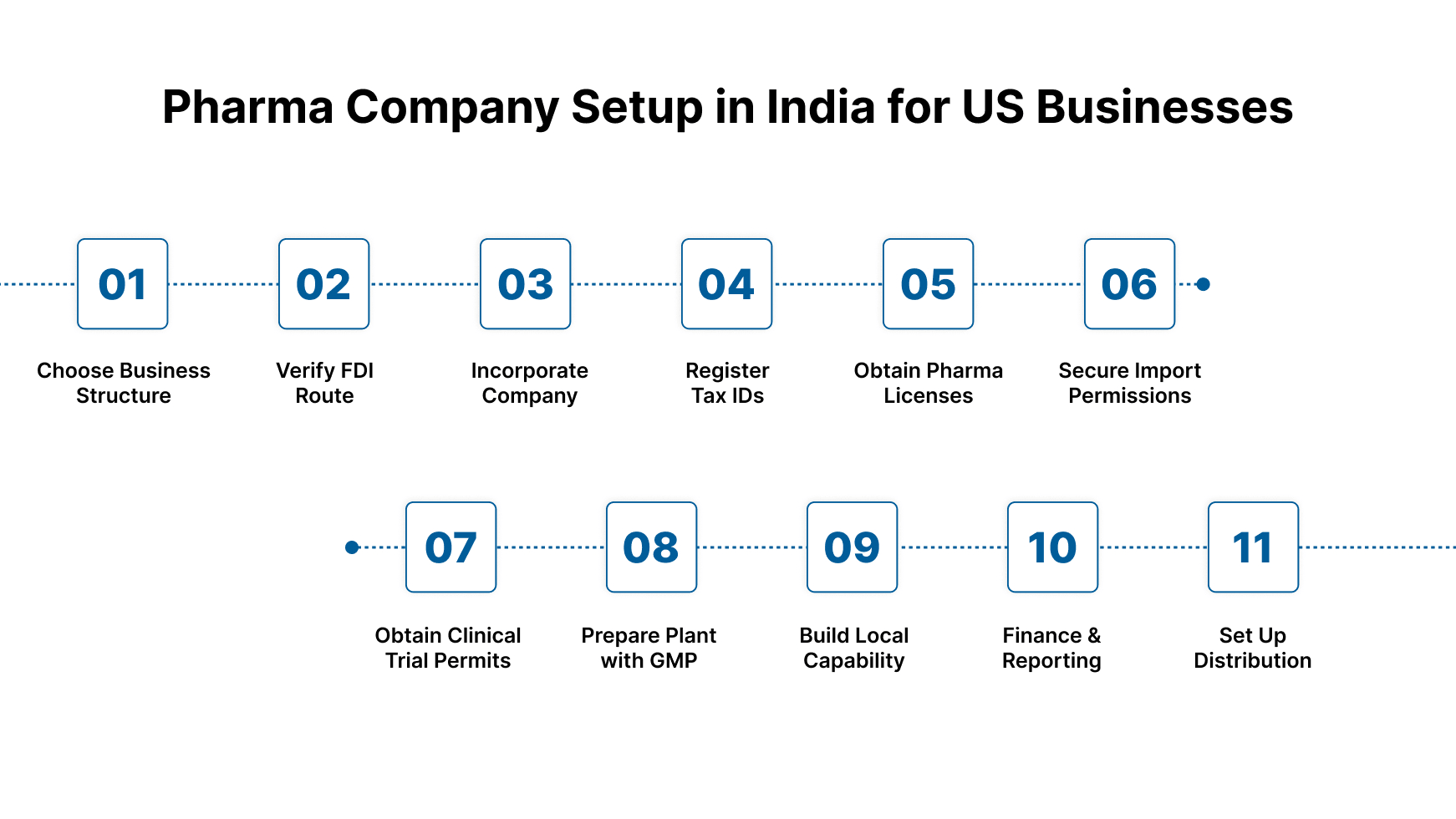
米国からインドに製薬会社を設立する方法に関するステップバイステップガイド
インドで製薬会社を設立するには、慎重な計画と規制基準の遵守が必要です。各ステップには、法律、財務、および業務上の異なるタスクが含まれます。手順と関係する権限がわかれば、プロセスはよりスムーズになります。
ステップ1:インドで製薬会社を設立する方法に適した事業構造を選択する
事業体の選択によって、統制、ライセンス経路、税務上の取り扱い、および投資家の準備が決まります。ほとんどの米国の創業者は、製薬会社として非公開有限会社または完全子会社を利用しています。LLPは小規模なサービス部門に適していますが、規制対象の製造にはほとんど適していません。海外の「オフィス」施設は存在しますが、製造には通常、インドに設立された会社が必要です。
提出する前に、次の簡単な比較を行ってください。
| 選択肢 |
外国資本比率 |
最適な用途 |
強み |
制限事項 |
| 株式会社(非公開会社) |
最大100%(FDI規制に準拠) |
将来の拡大を見据えた新規参入 |
投資家に優しい株式構造、明確なガバナンス、取締役の追加が容易 |
LLPよりも継続的なコンプライアンス負担が高い |
| 完全子会社 |
米国親会社が100%保有(FDI規範内) |
グループ報告を伴う完全なコントロール |
明確な移転価格制度、ブランドおよび品質管理の統一 |
厳格なFDI報告および監査が必要 |
| LLP(有限責任事業組合) |
外国パートナーの参加が可能 |
小規模なR&Dまたはサービス拠点 |
コンプライアンス負担が低く、利益配分が柔軟 |
多くの投資家に好まれず、製造ライセンスには不向き |
| 駐在員事務所/支店/プロジェクトオフィス |
RBIの許可ベース |
代表業務や限定的な活動 |
非製造目的で迅速に市場進出が可能 |
通常は製造が許可されず、活動範囲が限定的 |
提出前に準備するもの:
- 提案された会社名、製薬活動を対象とする主な目的、および登録された事務所証明。
- 将来の資金調達計画に合わせたKYC、デジタル署名、および資本構成をディレクターが担当します。
- 必要に応じて、医薬品の製造、輸入、販売を明示的に対象とする覚書および条項の草案。
期待できるアウトプット:
- 法人設立証明書、法人識別番号、およびPANとTANの自動割り当て。
- 銀行口座開設および主要管理職の任命に関する理事会決議
- 検査や監査の際に必要となる基本的な登録簿と議事録テンプレート。
実践的な例を以下に示します。
タブレットの製造と受託開発を計画している場合は、完全子会社を選択してください。API、製剤、輸入、販売、研究開発サポートをカバーできるよう、対象範囲を十分に広げてください。これにより、後で行を追加するときに修正する必要がなくなります。
初日から避けるべき一般的な落とし穴:
- 後で株式を調達したり、複数の製造ライセンスを申請したりする場合にLLPを選択します。
- API、FDF、または輸入および卸売業務を対象としない法人設立文書内の対象を絞り込みます。
- キャップテーブル計画をスキップすると、その後のFDI申請や移転価格文書化が複雑になる。
- 既存の製薬ブランドに近すぎる名前の選択は、反対意見や遅延を招きます。
ステップ 2: FDIルートの確認とオーナーシッププランの設定
インドに資金を持ち込む前に、製薬ベンチャーがどの外国直接投資(FDI)ルートに該当するかを確認してください。FDIの枠組みによって、自動的に投資できるかどうか、または事前に政府の承認が必要かどうかが決まります。オーナーシップ構造に誤りがあると、事業が遅れたり、事業が無効になったりする可能性があるため、このステップは非常に重要です。
医薬品に関するFDI規則:
- グリーンフィールドプロジェクト (新しい製造施設): 最大 100% 自動ルートでは外国人の所有が許可されています。ここでは政府の承認は必要ありません。
- ブラウンフィールドプロジェクト(既存のインドの製薬会社の買収または投資): 最大 74% 外国人の所有権は自動的に許可されます。74% を超える場合は、インド政府の承認を得る必要があります。
- 医療機器: 薬物とは別に治療すれば可能になる 100% 自動ルート下でのFDI。
準備すべきもの:
- 計画がグリーンフィールドかブラウンフィールドかを確認してください。たとえば、ハイデラバードに新工場を建設する場合、それは未解決のケースです。既存のインドの製薬会社の株式を取得する場合、その株式はブラウンフィールドになります。
- 法人設立の書類作成を始める前に、予定している株式保有額をこれらの上限に合わせてください。これにより、後で株式を再編する必要がなくなります。
- 取締役候補者、利益還元方針、内部報告項目を明記した明確な取締役会およびオーナーシップ・プランを設定します。
対処しなければならないコンプライアンス:
- すべてのFDI流入は、以下で報告する必要があります RBIの企業/SMFポータル 厳しい期限内(資金の受領には30日、株式の割当には60日)。
- 銀行証明書、外国送金証明書(FIRC)、および評価レポートはコンプライアンスパックの一部となります。
米国の創業者をつまずかせている落とし穴:
- 自動ルートか承認ルート内かを確認する前に、米国の親会社に株式を発行します。
- RBIの報告期限を逃すと、申請が複雑になり、追加の訴訟費用が発生します。
- 配当金の本国送金と移転価格を明確にしないで所有権を構築すると、後で税務効率に影響する可能性があります。
また読む: 医薬品セクターにおける外国直接投資 (FDI) 政策
ステップ3:会社を設立する
FDIルートとオーナーシッププランを確認したら、次のマイルストーンは製薬会社をインドに正式に設立することです。法人化することで法的な身分証明書が得られ、これがないとライセンスの申請、銀行口座の開設、課税登録ができなくなります。
完了する必要があるコアプロセス:
- デジタル署名 (DSC): 電子申告を可能にするには、すべての取締役のDSCを入手してください。
- 取締役識別番号 (DIN):各候補取締役の設立フォームから申請してください。
- Spice+ ウェブフォーム: 社名予約についてはパートAを、企業省のポータルに組み込む場合はパートBを提出してください。これには、資本、登録事務所、および事業目的の詳細が含まれます(対象範囲に含まれる場合は、必ず「医薬品の製造、輸入、販売」と明記してください)。
- リンクされた申告書:同じプロセスを使用して PAN、TAN、およびオプションで EPFO/ESIC 登録を取得します。
準備が必要な書類:
- 取締役および株主の身元および住所証明。
- 登録事務所の証明(リース証書または公共料金請求書)。
- 製薬事業を対象とする定款覚書(MOA)および定款(AOA)の草案。
- 取締役からの同意書および会社法に基づく失格ではない旨の宣言
最後に受け取るもの:
- 法人識別番号(CIN)付きの法人設立証明書。
- PAN と TAN は、承認時に自動的に割り当てられます。
- 新会社の銀行口座認証。
米国の創設者のための実践例:
米国の親会社が 100% 株主である場合は、MOAに加入者として記載します。親権者は、権限のある代理人を通じてデジタル署名を行います。これにより、オーナーシップの証跡が明確になり、次の段階でのFDI報告が容易になります。
避けるべき一般的な落とし穴:
- MOA/AOAオブジェクトの作成範囲が狭すぎる(例:製造のみを対象とし、輸入、販売、研究開発は対象としない)。これにより、後で修正を余儀なくされます。
- 不完全な登録事務所証明書を提出すると、MCAによる再提出のきっかけとなることがよくあります。
- 少なくとも1人の取締役がインドに居住している必要がある(会計年度に少なくとも182日間滞在)という要件を見落としている。
ステップ 4: 税および法定IDを登録する
設立後、製薬会社が合法的に事業を行うには、税務および法定登録が必要です。これらの ID は、インドにおける財務および規制活動の基盤となります。ID がないと、顧客への請求や税金の控除、商品の輸出入はできません。
保護する必要のある主な登録:
- 永久口座番号 (PAN): 会社の税務上の身分証明書として機能します。
- 税額控除および徴収口座番号 (TAN): 給与、契約者への支払い、またはその他の特定経費から源泉徴収(TDS)を控除する場合は必須です。
- 物品サービス税 (GST) 登録: 商品やサービスの提供を計画している場合に必要です。製薬会社は医薬品を製造または販売しているため、ほとんどの場合、GSTが必要です。
- インポーターエクスポーターコード (IEC): API をインポートするか、製剤をエクスポートする場合は必須です。
プロセスには以下が含まれます。
- PANとTANは通常、Spice+フォームを介して組み込み時に自動的に割り当てられます。法人設立証明書を受け取ったら、それらが有効であることを確認してください。
- GSTポータルでGSTを申請してください。勤務先住所の証明、PAN、および取締役の詳細が必要です。
- 基本的な企業書類と銀行情報を添えて、外国貿易総局(DGFT)ポータルでIECを申請してください。
クイックリファレンス用のコンプライアンス表:
| 登録 |
目的 |
更新 |
備考 |
| PAN |
主要な納税者識別番号 |
永久 |
すべての税務申告に使用 |
| TAN |
TDS(源泉徴収)用 |
永久 |
TDSの納付および申告に必要 |
| GST |
商品・サービスに対する間接税 |
年次申告 |
医薬品販売に必要 |
| IEC |
輸出入の許可 |
永久 |
APIおよび輸出に必須 |
フォローできる例:
米国企業がジェネリック医薬品を製造するためにインドに子会社を設立する場合、設立時に自動的にPANとTANが必要になり、売上税を請求するにはGSTが必要になり、未加工のAPIを海外から輸入するにはIECが必要になります。
避けるべき一般的な落とし穴:
- GSTを早めに有効化し忘れると、有効な請求書を発行できなくなります。
- 最初の出荷前にIECを申請しなかった場合、税関での遅延につながります。
- PAN と TAN はオプションとして扱います。経理を外部委託する場合でも、両方とも必須です。
ステップ5:インドで製薬会社を設立する方法に必要な製薬ライセンスを取得する
会社が設立され、税務登録が行われたら、次の重要なステップは、医薬品の合法的な製造、販売、流通を可能にするライセンスを確保することです。インドでは、ライセンスに関する責任は以下に分かれています。 中央医薬品標準管理機構 (CDSCO) 国レベルで、州レベルでは州食品医薬品局(FDA)。
扱うべきコアライセンスと承認:
- 製造ライセンス:
- 州FDAによって発行されました。
- を使用して申し込む フォーム 25 ほとんどの薬と フォーム 28 無菌または非経口製品(注射剤などのスケジュールCおよびC1薬)を製造している場合。
- 卸売および小売ライセンス:
- フォーム 20/21 小売販売用。
- フォーム 20B/21B 卸売流通用。
- これらは州FDAによって発行され、通常は登録薬剤師の任命が必要です。
- 中央承認 (CDSCO/DCGI):
- 新薬、生物製剤、ワクチン、および特定の高リスクカテゴリーに必要です。
- すべての申請書は以下を通じて提出されます スガムポータル、これはCDSCOのオンラインライセンスプラットフォームです。
事前に準備しておくべきこと
- 資格のある技術スタッフ: 必要な資格を持つ薬剤師または化学者を雇用し、ライセンス機関に「有能な技術スタッフ」として登録する必要があります。
- サイトドキュメンテーション: 工場のレイアウト、設備の詳細、スケジュールMに基づく適正製造基準(GMP)への準拠の証明
- 品質保証文書:標準運用手順(SOP)、品質管理措置、および記録管理システム。
フォローできる例:
グジャラート州でジェネリック経口錠剤を製造することから始めたい場合は、 フォーム 25 製造ライセンス 州のFDAから。また、資格のある技術スタッフがいることを証明し、スケジュールMの要件に準拠していることを示すプラントレイアウトを提出する必要があります。
避けるべき一般的な落とし穴:
- 製造予定の薬剤の種類に基づいて、間違った申請書(様式28の代わりに様式25など)を申請すること。
- 有能な技術スタッフを雇用していない、または書類を作成していない。これが、申請が却下される標準的な理由です。
- 不完全なプラントまたは品質文書を提出すると、承認プロセスが大幅に遅くなります。
ステップ 6: 原薬と完成薬の輸入許可の確保
インドでの製薬事業計画にインド国外からの医薬品有効成分(API)、原材料、または完成した製剤の輸入が含まれる場合は、特定の輸入許可を取得する必要があります。これらは中央医薬品標準管理機構 (CDSCO) によって一元的に処理されます。これらがないと、貨物は税関で保管され、規制上の罰則を受けるリスクがあります。
知っておくべきコアインポートライセンス:
- フォーム 10: 制限対象カテゴリーに記載されていない医薬品の標準輸入許可証。
- フォーム 10A: スケジュールXの医薬品(麻薬および向精神薬)専用の輸入ライセンス。
- フォーム 41 (登録証明書):海外メーカーおよびその施設向けに発行されました。海外のサイトと製品がインドの規制基準を満たしていることが確認されました。
- BA/BE 学習教材: バイオアベイラビリティ/生物学的同等性試験のために少量のバッチを輸入する場合は、特別な許可が必要です。
実行する必要がある手順:
- フォーム10または10Aを申請する前に、SUGAMポータルを通じて外国の製造拠点と特定の製品をCDSCOに登録してください。
- 原産国のGMP証明書、試験プロトコル、安定性データを含む完全な書類を提出してください。
- 輸入と流通の法的責任を負うインドの認定代理人または子会社を任命してください。
フォローできる例:
米国の施設から抗生物質錠剤のAPIをインポートする予定の場合、米国のサイトは最初にForm 41の登録証明書を受け取る必要があります。そうして初めて、インドの子会社はそれらの特定の API を対象とする Form 10 輸入ライセンスを申請できます。
避けるべき一般的な落とし穴:
- 最初にForm 41で製造元を登録せずに輸入を試みた。税関はこのような貨物の通関手続きは行いません。
- 卸売ライセンスの保管要件(生物製剤のコールドチェーンなど)を見落とすと、ライセンスの一時停止につながる可能性があります。
- 適切な権限を持つインドの代理人を任命していない。これは、インドに子会社を持たない海外メーカーには必須です。
ステップ 7: 臨床試験と新薬の許可を取得 (該当する場合)
事業計画にインド市場への新薬の導入または臨床試験の実施が含まれる場合は、以下の承認を得る必要があります。 新薬および臨床試験規則、2019年(NDCTR)。これらの規則は、治験施設の登録から倫理委員会の承認まですべてを規定し、インド基準と国際基準の両方への準拠を保証します。
これがあなたに当てはまる状況:
- インドでまだ承認されていない新しい化学薬品または生物製剤の発売
- ジェネリック版のバイオアベイラビリティ/生物学的同等性(BA/BE)研究の実施。
- 以前に承認されていない固定用量の併用(FDC)を紹介します。
必要なコア権限と登録:
- 臨床試験申請書は中央ライセンス局に提出されました(CDSCOを通じて)。
- 使用するすべてのトライアルサイトについて、倫理委員会をCDSCOに登録します。
- 治験実施施設の承認により、病院または施設がインフラと人員の基準を満たしていることを確認できます。
- BA/BE試験の承認(ジェネリック医薬品と参照薬との比較試験を行う場合)
準備しなければならないもの:
- 前臨床および臨床データ、製造の詳細、治験責任医師向けパンフレットを含む書類一式。
- 倫理委員会からの承認書および治験責任医師との合意
- グッドクリニカルプラクティス(GCP)ガイドラインの遵守、およびモニタリングと報告を対象とするSOPの遵守
フォローできる例:
インドでジェネリック心臓血管薬の生物学的同等性試験を実施する予定の場合は、治験実施施設の承認、倫理委員会への登録、およびBA/BEプロトコルのCDSCO認可が必要です。これがないと、生成したデータは市場承認の対象にはなりません。
避けるべき一般的な落とし穴:
- 中央ライセンス局の承認を得ずに試験を開始すると、試験全体が無効になります。
- CDSCOに登録されていない倫理委員会を利用すると、試験が中断されるリスクがあります。
- CDSCOは現場での検査を省き、GCPの遵守状況について試験センターを頻繁に監査します。
ステップ8:GMPコンプライアンスと環境クリアランスを遵守してプラントを準備する
インドでの製造を計画する場合、施設は以下の適正製造基準(GMP)を満たしている必要があります 医薬品および化粧品規則のスケジュールM。これに加えて、事業を開始する前に州当局から環境許可を取得する必要もあります。
GMPコンプライアンスで確立する必要があるもの:
- インフラストラクチャー標準:生産、品質管理ラボ、保管、および包装用の分離エリア。それぞれが相互汚染を防ぐように設計する必要があります。
- ドキュメンテーションと SOP: 検査官がいつでも監査できるバッチ製造記録、品質保証プロトコル、および変更管理手順。
- 品質システム: 工程内の品質チェック、安定性試験設備、設備とプロセスの検証。
スキップできない環境権限:
- 設立への同意 (CTE): 機械を設置したり建設を開始したりする前に、州公害防止委員会によって発行されます。
- 運営への同意 (CTO): 生産開始前に、工場が水・大気法に基づく基準を満たしていることを確認する必要があります。
- 有害廃棄物認可: 工場で特別な廃棄が必要な溶剤、化学薬品、または副産物を処理している場合。
フォローできる例:
ハイデラバードにグリーンフィールドプラントを設立する米国企業は、まずCTEを取得し、次にプラントレイアウト、排出制御、廃棄物管理システムの証明を提出する必要があります。設置後は、商業用バッチを立ち上げる前にCTOを確保する必要があります。それと並行して、州FDAによる検査に合格するには、プラント設計がスケジュールMに従わなければなりません。
避けるべき一般的な落とし穴:
- 設立の同意を受ける前に機器を購入または設置すると、作業停止命令が出る可能性があります。
- 溶剤回収システムや有害廃棄物システムは無視されます。これらは検査中にフラグが立てられることがよくあります。
- GMP SOPは紙の上でのみ作成し、日常業務と整合させていないため、コンプライアンス違反の指摘につながる。
ステップ 9: ローカル能力と運用統制の構築
施設が認可を受け、規制に準拠したら、製薬業務を管理するための適切な人材とシステムが社内に揃っていることを証明する必要があります。規制当局は、品質とコンプライアンスを維持するための有能なスタッフと強固な内部統制の有無に重点を置いています。
満たす必要のある中核的な人員配置要件:
- 薬剤師と技術スタッフ: 医薬品および化粧品規則で義務付けられているように、薬局または化学の資格を持つ「有能な技術スタッフ」を任命する必要があります。彼らの名前と資格はライセンスに記録されています。
- 品質保証 (QA) および品質管理 (QC) リーダー: バッチ記録、偏差、安定性調査、製品リコールを監督する必要があります。
- 規制コンプライアンス責任者: CDSCO、州食品医薬品局 (FDA) への提出を処理し、記録が検査準備が整っていることを確認します。
実装すべき運用システム:
- 標準運用手順 (SOP): クリーニング検証からバッチリリースまでのすべてのアクティビティを、バージョン管理とスタッフトレーニング記録でカバーします。
- 文書化と記録管理:バッチ製造記録、逸脱ログ、および是正措置/予防処置(CAPA)レポートを管理します。
- サプライヤーと委託製造業者の監督: 監査を実施してベンダーを認定し、原材料と外注作業がGMP基準を満たしていることを確認します。
フォローできる例:
インドでジェネリック腫瘍治療薬を製造する米国子会社では、通常、登録薬剤師を技術責任者、検査室運営のQCマネージャー、リリースプロセスのQA責任者を雇用しています。このチームはSOPを維持し、トレーニングを監督し、インド当局と米国FDAの監査双方の検査準備状況を確認しています。
避けるべき一般的な落とし穴:
- 資格のない技術スタッフを雇用すると、検査中に免許が停止される可能性があります。
- SOPを日常業務に組み込むのではなく、書類として扱うことで、検査官がすぐに気付くようになります。
- ベンダーの資格は無視します。未確認のサプライヤーが標準以下の原材料を提供した場合、あなたは責任を問われます。
ステップ10: 財務、国境を越えた報告、継続的な申請
業務を開始すると、コンプライアンスの負担は財務報告や規制報告に大きく移ります。企業は外国直接投資 (FDI) を伴うため、規制当局は国境を越えた資金流入、株式保有、継続的な税務申告を綿密に追跡します。
FDIとRBIの報告義務:
- 外国送金証明書 (FIRC): 米国の親会社から資金が届くたびに、銀行からこれらを回収してください。
- FC-GPR ファイリング: 割当から30日以内に、RBIのFIRMS/SMFポータルで株式割当を報告してください。
- FLA の年間リターン: 毎年、外国負債と資産をRBIに開示します。
税務および法定申告書には以下を必ず把握しておく必要があります。
- 法人所得税: 監査済み財務諸表とともに年次報告書を提出してください。米国の親会社と取引を行う場合は、移転価格に関する書類が不可欠です。
- GST申告書: 売上と購入の月次または四半期ごとの返品と年次調整。
- TDS コンプライアンス: 給与、請負業者の支払い、および仕入先請求書の税金を源泉徴収し、四半期ごとにTDS申告書を提出します。
導入すべき内部統制:
- すべての請求書、国境を越えた契約、および会社間契約の監査記録を維持します。
- エラーを回避するには、GSTおよびTDSモジュールと統合された会計システムを使用してください。
- スケジュール 内部監査 法定監査の前に不一致を早期に発見します。
フォローできる例:
インドの子会社が製造工場を設立するために100万ドルのFDIを受け取った場合は、米国の親会社に株式を発行してから30日以内にFC-GPRを申請する必要があります。その後、国内で医薬品を販売する場合、GST請求書を毎月提出する必要があります。保護者への委託研究サービスも提供する場合は、 移転価格 申告書は所得税申告書と一緒に提出する必要があります。
避けるべき一般的な落とし穴:
- RBIのFDI報告期限の30日または60日を逃すと、罰則付きの複利申請を余儀なくされます。
- 適切なベンチマークを行わずに会社間取引を記録すると、移転価格紛争につながります。
- GSTインプットクレジットを仕入先請求書と照合できなかったため、監査中にクレジットが認められなくなった。
ステップ11:インドで製薬会社を設立する方法のための流通および二次事業の設定
製造または輸入業務の準備が整ったら、最後のステップは流通チャネルと二次販売チャネルを確立することです。これにより、製品が病院、薬局、卸売業者に法的にかつ厳格なコンプライアンスの下で確実に届きます。
確保する必要のあるライセンス:
- 卸売ライセンス (フォーム 20B および 21B): 病院、診療所、または小規模小売店に配布する場合に必要です。
- リテールライセンス (フォーム 20 および 21): 薬局を通じて消費者に直接販売する場合に必要です。
- ストレージの承認: 生物製剤またはワクチンのコールドチェーン施設は、州のFDAによる検査と承認が必要です。
オペレーションの構成方法:
- 倉庫: 独自の施設を建設するか、認定されたロジスティクスプロバイダーと契約してください。倉庫は、州FDAが規定する保管、セキュリティ、および衛生条件を満たしている必要があります。
- 契約分配: サードパーティのディストリビューターを任命することもできますが、ライセンスと契約では役割と説明責任を明確に定義する必要があります。
- 技術サポート: 在庫管理システムとバッチ追跡システムは、監査証跡を維持し、必要に応じて製品のリコールを可能にするために不可欠です。
フォローできる例:
米国の顧客向けに完成したジェネリック医薬品を輸入し、インド国内で販売する場合、子会社には卸売ライセンス(20B/21B)が必要です。ムンバイの規制に準拠した倉庫で業務を行い、ワクチンの取り扱いには冷蔵トラックの販売業者を指名できます。
避けるべき一般的な落とし穴:
- ライセンスのないサードパーティ倉庫を使用すると、卸売ライセンスが無効になります。
- 温度に敏感な医薬品のコールドチェーンの完全性が維持されないため、規制措置が取られることになる。
- 検査官が監査中に確認する販売記録と請求書を適切に保存していない。
また読む: 米国からインドのバイオテクノロジー企業を始める方法
With VJM Global, entry to India is made simple; company registration, compliance, and ongoing support in one place.
設定手順に従って、業務と規制当局の承認に直接影響するコンプライアンス分野に焦点を当てる必要があります。
米国の製薬企業家にとってのコンプライアンスに関する主な考慮事項
インドの医薬品市場に進出する場合、長期的な成功は厳格なコンプライアンス要件を満たすことにかかっています。当局は企業が機密データを保護し、製造基準と試験基準を満たし、国内外の規制を遵守することを期待しています。
1。データ保護と機密保持
- 臨床試験またはBA/BE研究中に患者データを扱う場合は、インドの規定に従う必要があります 情報技術法 および機密個人データに関する規則。
- 委託研究機関(CRO)または第三者ベンダーとの契約には、秘密保持契約と機密保持条項を含める必要があります。
- 米国企業にとっては、HIPAAまたはGDPR基準に準拠することがベストプラクティスです。これは、世界の規制当局が治験記録を照合することが多いためです。
2。製品テスト要件
- 製造されたすべてのバッチは、リリース前に安定性試験と工程内品質管理を受ける必要があります。
- インドに輸入される医薬品は、市場に出る前に政府が承認した研究所で試験を受ける必要があります。
- バッチリリース文書、逸脱ログ、および是正措置/予防措置(CAPA)記録を担当する品質保証(QA)ユニットを設立する必要があります。
3。インポートとエクスポートのルール
- 原薬または完成薬は、有効なフォーム10またはフォーム10Aのライセンスと、海外メーカーのフォーム41の登録証明書がないと輸入できません。
- 輸出業者は以下を取得する必要があります インポートエクスポートコード (IEC) 特にスケジュールXに基づく規制薬物については、税関の報告を順守してください。
- 出荷書類には、コールドチェーンおよび危険物の取り扱い要件への準拠を示す必要があります。
4。臨床試験の承認
- 新薬またはBA/BE研究を含む試験には、2019年の新薬および臨床試験規則に基づく中央ライセンス局(CDSCO)の許可が必要です。
- 各トライアルサイトは承認され、登録された倫理委員会にリンクされている必要があります。
- 米国のスポンサーは、申請を管理し、インドのGCP基準と米国FDA基準の両方に準拠していることを確認するために、インドの子会社またはCROを任命することがよくあります。
5。スケジュール M の製造基準
- 工場は、施設、設備、文書、および品質システムの適正製造基準(GMP)を定めた医薬品化粧品規則のスケジュールMに準拠する必要があります。
- 検査員は、検証済みのプロセス、資格のある人材、完了したSOPを確認することを期待しています。
- スケジュールMに従わないと、製造許可が一時停止される可能性があり、違反が繰り返されると、米国への輸出能力に影響が出る可能性があります。
主要なコンプライアンス分野のクイックスキャン表:
| コンプライアンス分野 |
管轄機関 |
主な要件 |
影響例 |
| データ保護 |
IT法(インド)、HIPAA/GDPR(国際) |
患者データの保護 |
CRO契約にはNDAの締結が必要 |
| 製品試験 |
CDSCO、州FDA |
バッチ品質管理、安定性データ |
不合格バッチは出荷不可 |
| 輸出入 |
CDSCO、DGFT、税関 |
有効な申請書類、IECコード |
承認なしでは港で貨物が保留される |
| 臨床試験 |
CDSCO、倫理委員会 |
試験および施設の承認 |
承認がない場合、試験データは無効 |
| 製造(GMP) |
CDSCO、州FDA |
スケジュールMへの準拠 |
GMP違反によりライセンス停止 |
また読む: インドでの事業立ち上げ:起業家にとって不可欠なステップ
このようなコンプライアンス要件を満たすことは容易ではありません。そこで、VJM Globalは専門家によるガイダンスと継続的なサポートを提供します。
VJM Globalがインドに製薬会社を開設する米国企業をどのようにサポートしているか
インドで製薬会社を設立するには、ライセンスや工場の承認だけでは不十分です。また、法人設立、コンプライアンス、継続的な財務業務を管理できる信頼できるパートナーも必要です。このような場面があります。 VMグローバル 米国の起業家にとって極めて重要な役割を果たしています。
VJM Globalで得られる専門サポート:
- 会社の設立と登録: 法人設立、憲章文書の作成、PAN、TAN、GSTなどの法定IDの確保を支援します。
- 連邦緊急事態管理局とRBIのコンプライアンス: 罰則を回避するためのFDI流入額、FC-GPR申請、および継続的な外国為替コンプライアンスに関するタイムリーな報告
- GSTと税務コンプライアンス: 登録、毎月の返品、照合により、インドの規制に沿った円滑な運営が保証されます。
- 会計アウトソーシング: 帳簿管理、財務報告、給与計算の両方について研修を受けたチームがエンドツーエンドで対応 米国会計基準とインド基準。
- 監査サポート: 監査スケジュールの作成、調整、統制テストにより、法定監査または内部監査時の作業負荷を軽減します。
米国の起業家としてこれが重要な理由:
- インドに大規模な財務チームやコンプライアンスチームを組織しなくても、長期的にコンプライアンスを維持できます。
- 会計や規制業務をアウトソーシングすることで、管理費の40~ 50% を節約でき、それを研究開発、製造のアップグレード、または臨床試験に振り向けることができます。
- インド事業が米国とインドの両方の規制上の期待に応えていると確信できるので、監査人と投資家は安心できます。
インドで製薬会社を設立する方法を検討している場合、VJM Globalは会社の設立、アウトソーシング、およびビジネスのコンプライアンス面を処理する専門知識を提供するので、お客様は成長に集中できます。
Set up your Indian entity faster with VJM Global’s end-to-end business registration services.
結論
インドの製薬セクターへの進出により、成長市場へのアクセス、コスト削減、熟練労働力の確保が可能になります。成功するかどうかは、適切な組織を選択し、ライセンスを確保し、規制や財務上の要件を完全に遵守できるかどうかにかかっています。適切なサポートがあれば、プロセスは管理しやすく、費用対効果も高くなります。
適切なコンプライアンスとコストサポートを備えた製薬会社をインドに設立する準備はできていますか? VMグローバル 米国の起業家が企業登録、FEMA/RBI報告、GST、会計アウトソーシング、監査サポートを処理するのを支援し、経費を削減しながら業務のコンプライアンスを維持できるようにします。今すぐVJM Globalと提携して、自信を持って事業拡大の旅を始めましょう。
自信とコンプライアンスをもって製薬事業をインドに拡大する準備はできていますか?米国会計基準とインドの規制に関するVJM Globalの二重の専門知識により、企業は管理費を最大 50% 削減できます。 VJM Globalとの面談を今すぐスケジュールしてください コンプライアンスに準拠したコスト効率の高いセットアップへの第一歩を踏み出しましょう。
よくある質問
Q: インドで製薬会社を設立するために必要な最低投資額はいくらですか?
A: 必要な投資は規模によって異なります。小規模な調合ユニットは20万ドル未満から始めることができますが、大規模なプラントでは数百万ドルの予算が必要です。
Q: 独自の製造施設を建設せずにインドで事業を開始できますか?
A: はい。FDAの承認を受けた工場を持つ委託製造組織を利用できるため、多額の資本支出なしに迅速に市場に参入できます。
Q: 輸入された API がインドの品質基準を満たしていることを確認するにはどうすればよいですか?
A: インポートされた API は CDSCO に登録する必要があります。市場にリリースする前に、政府が承認したラボでのテストが義務付けられています。
Q: コンプライアンス申請を処理するには、現地のパートナーが必要ですか?
A: 必ずしもそうではありません。完全子会社でもコンプライアンスを管理できますが、多くの米国企業は信頼できるインド企業に申告を外部委託しています。
Q: 法人化およびライセンス取得後、どのくらい早く販売を開始できますか?
A: 法人化には2〜3週間かかる場合があります。ライセンス、輸入許可、およびGST登録により、期間が6〜12か月に延長されることがよくあります。
Q: VJM Globalは米国とインドの両方の会計要件を同時に管理できますか?
A: はい。VJM Globalのチームは、米国会計基準とインド基準に関する研修を受けており、法域全体で統一された報告と一貫したコンプライアンスを確保しています。


%20(13).webp)